Les cardiopathies pédiatriques non cyanogènes
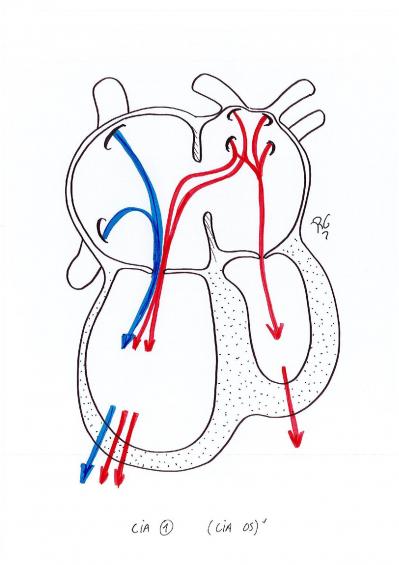
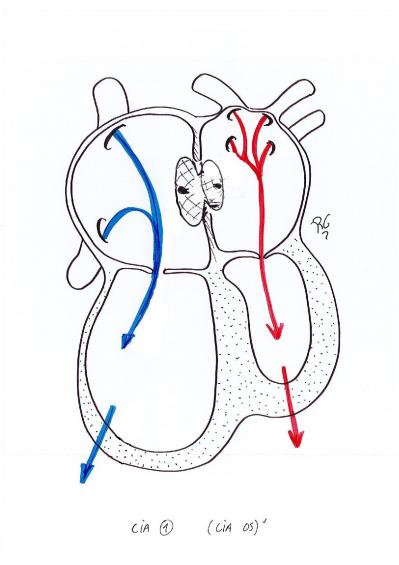
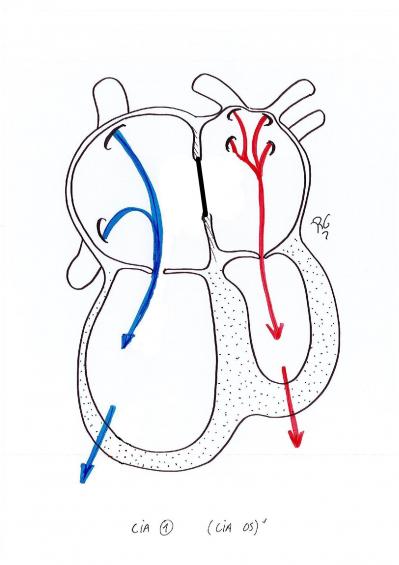
Le traitement consiste à fermer cette communication soit chirurgicalement à l’aide d’un patch soit par voie percutanée à l’aide d’une prothèse.
Communication inter ventriculaire : CIV
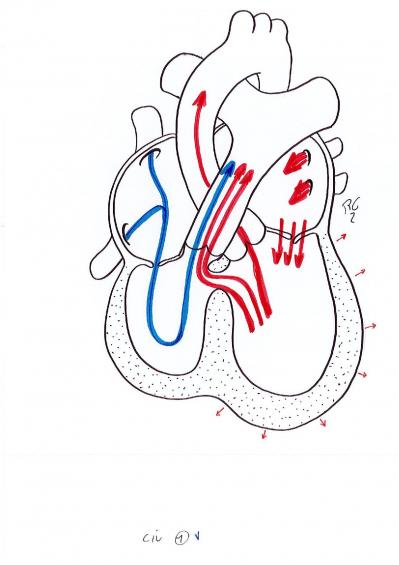
Le traitement consiste à fermer cette communication le plus souvent par voie chirurgicale.
Avant la chirurgie en cas de CIV importante, une transfusion peut être réalisée afin d’augmenter la viscosité du sang et limiter l’hyperdébit et ses conséquences délétères sur la santé de l’enfant. Une prophylaxie des bronchiolites à VRS (injection d’immunoglobulines) chez les nourrissons de <1 an est très importante et permet d’éviter une partie des hospitalisations dues à ces viroses respiratoires.
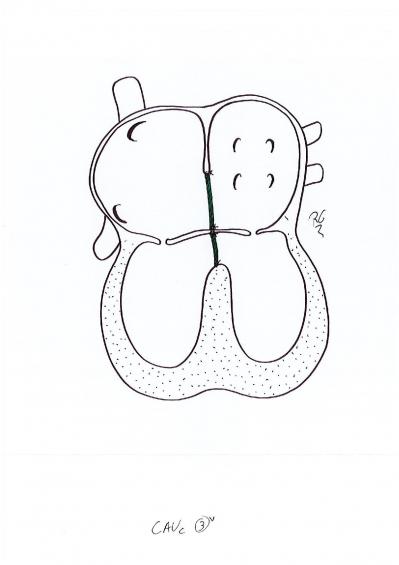
2- Canal atrio-ventriculaire : CAV
C’est une anomalie congénitale avec la persistance d’une valve auriculo ventriculaire unique associées à une CIA et CIV. Cette malformation est souvent associée à la trisomie 21 ou autre syndromes. Elle est souvent diagnostiquée en anténatal.
Les symptômes sont similaires à la CIV.
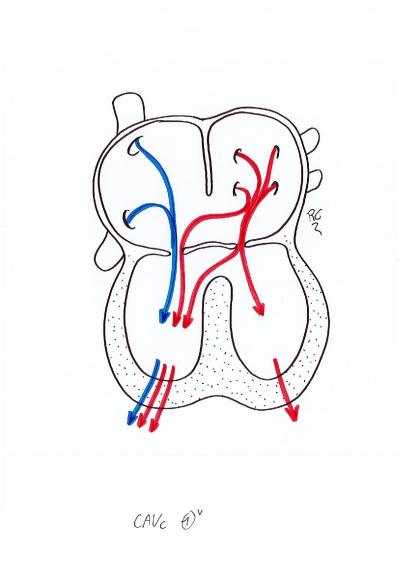
Le traitement consiste en une chirurgie à cœur ouvert entre 3 et 6 mois.
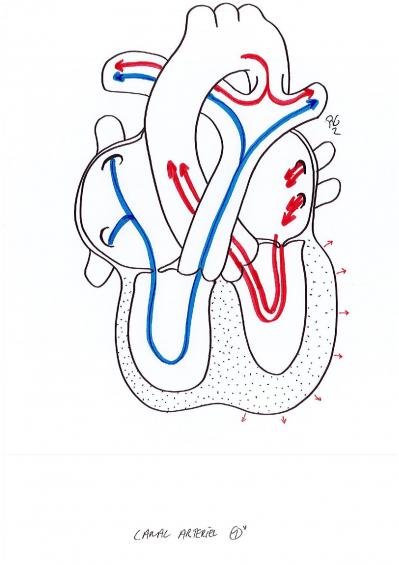
Lorsque le canal artériel est très large (égalisation des pressions entre l’aorte et l’artère pulmonaire) ou si l’hyperdébit pulmonaire est important, on peut le fermer par cathétérisme (pose d’une prothèse) ou par chirurgie s’il est trop large (ligature section).
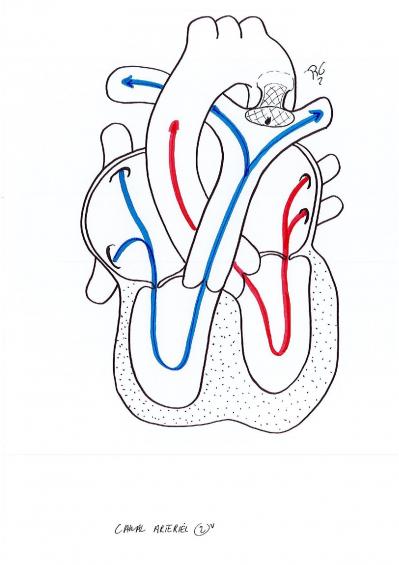

4- Obstacle à l’éjection du cœur gauche ou droit
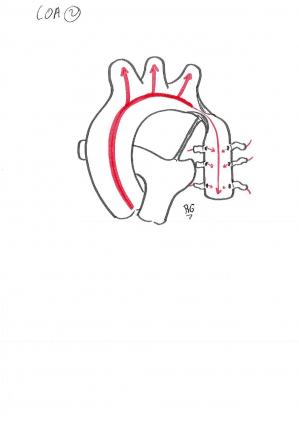
La coarctation de l’aorte
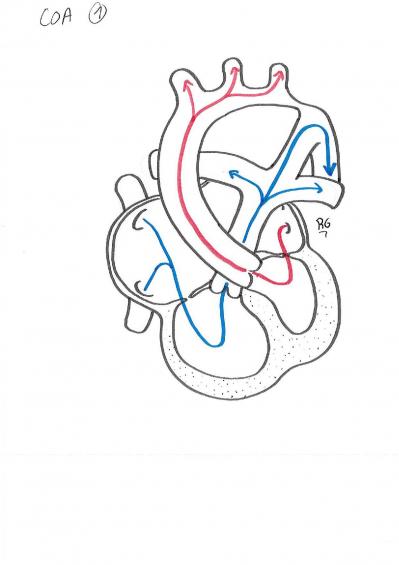
En pré-opératoire pour les nouveaux nés symptomatique, une perfusion de prostaglandine est administrée. Elle permet de maintenir le canal artériel large afin de limiter les effets de la coarctation, en attendant la chirurgie.
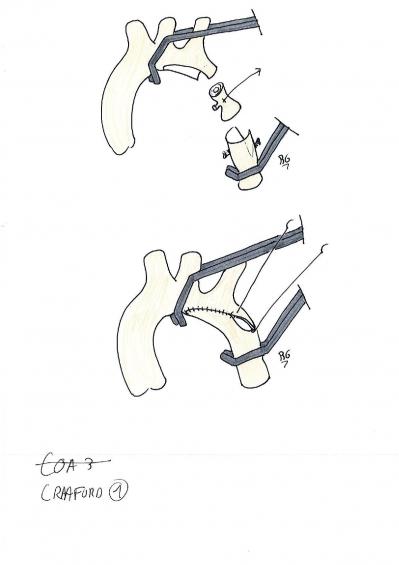
Le traitement consiste en une chirurgie de plastie de l’aorte ou un traitement par cathétérisme chez les enfants plus âgé ou les adultes (dilatation et pose de stent).
Le rétrécissement aortique
C’est un obstacle au niveau de la valve aortique qui constitue un obstacle à l’éjection du ventricule gauche.
Il est le plus souvent symptomatique chez les plus petits enfants (nouveaux nés ou nourrissons).
Chez les plus grands enfants, il y a le plus souvent pas de symptômes sauf lorsqu’il devient important (notamment à la puberté). Après plusieurs années, selon l’évolution du rétrécissement, le patient peut faire des syncopes, des dyspnées d’effort, de l’insuffisance cardiaque et/ou des troubles du rythme.
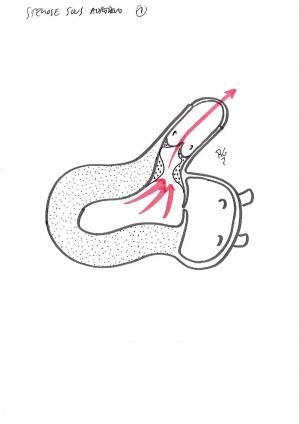
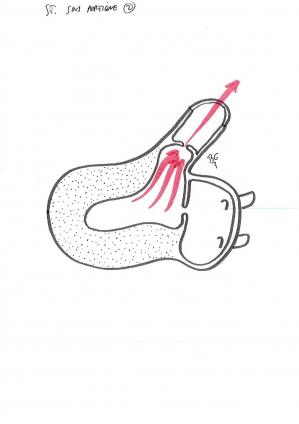
On traite les patients symptomatiques par chirurgie cardiaque (plastie ou un remplacement valvulaire) ou par cathétérisme cardiaque (dilatation par ballonnet).
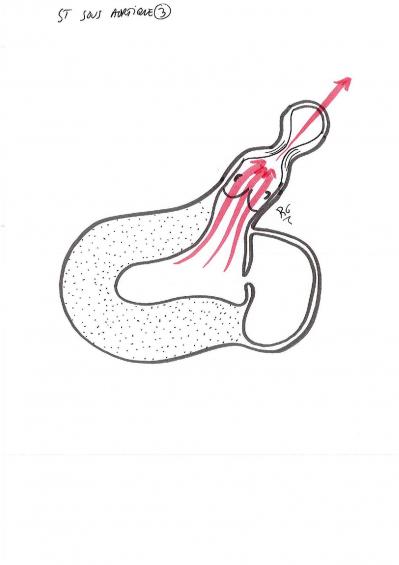
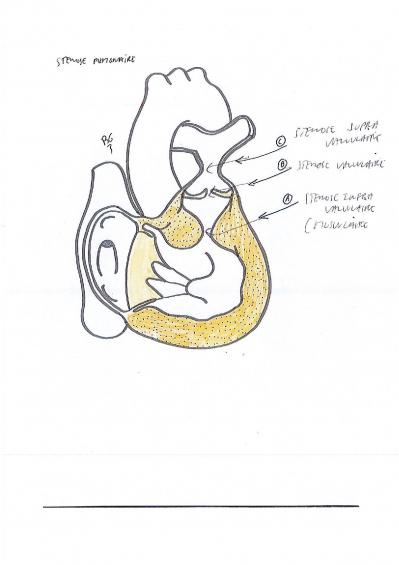
La sténose pulmonaire